Table of Contents
1. Introduction
RPF - Protein NMR structure quality assessment tool
RPF uses a novel, rapid, and simple approach for calculating global NMR structure quality scores. This program calculates RECALL, PRECISION, and F-MEASURE (RPF) scores assessing how well the query 3D structure(s) fit to the experimental NOESY peak list and resonance assignment data. RPF scores quickly assess the goodness-of-fit of the query structure(s) to these experimental data, and can be used as a guide for further structure refinements. RPF also calculates discrimination power (DP) scores, which estimate the difference in F-MEASURE scores between the query structure and "random coil" structures, as an indictor of the correctness of the overall fold. The program is useful for quality of control protein NMR structures determined by automated or manual methods.
2. Web-Server
RPF Web-Server
2.1 Input Files
Input Files for use with the RPF Web-Server
In order to use the RPF Web-Server, you must enter your the protein name, PDB file, BMRB file, and NMR peaklists. The BMRB file should be in 2.x or 3.x format, and the peaklists should be in Sparky or Xeasy format.
Services such as the FormatConverter server on the WeNMR website can be used to perform the conversion from other formats not supported by the RPF server.
If the experiment was performed in d20 solution, click the d20 checkbox for the appropriate peaklist. You can also specify a water suppression range in ppm-ppm format, eg. "4.6-4.9" to exclude this region for precision violation calculation.
Download sample input files
Limitations for analyzing larger size proteins and homodimeric proteins
For larger size proteins (e.g. > 200 a.a.), it is often necessary to use perdeuterated samples for structure determination. The RPF program can handle validation of protein structures using data from such perdeuterated protein samples, by excluding the deuterated atoms from the chemical shift assignment table. The computed RPF score provides useful measures of how good the data fits with the structure. However, the correlation between the RPF/DP scores and the structure accuracy is not as high as with fully-protonated proteins, because data from perdeuterated proteins is much more sparse. Additional test data sets are needed to assess the best way to use the DP score for data obtained on perdeuterated proteins.
The RPF program can also analyze homodimeric proteins. This requires the user to first combine the two identical chains into a single chain with different residue index. A new chemical shift assignment table can be created based on the resonance assignments from the two identical chains, and match with the combined single chain residue index. We suspect that an accurate highly-degenerate homodimeric protein structure will require a higher DP score cutoff than a protein structure with less degenerate resonance frequencies.
Services such as the FormatConverter server on the WeNMR website can be used to perform the conversion from other formats not supported by the RPF server.
If the experiment was performed in d20 solution, click the d20 checkbox for the appropriate peaklist. You can also specify a water suppression range in ppm-ppm format, eg. "4.6-4.9" to exclude this region for precision violation calculation.
Download sample input files
Limitations for analyzing larger size proteins and homodimeric proteins
For larger size proteins (e.g. > 200 a.a.), it is often necessary to use perdeuterated samples for structure determination. The RPF program can handle validation of protein structures using data from such perdeuterated protein samples, by excluding the deuterated atoms from the chemical shift assignment table. The computed RPF score provides useful measures of how good the data fits with the structure. However, the correlation between the RPF/DP scores and the structure accuracy is not as high as with fully-protonated proteins, because data from perdeuterated proteins is much more sparse. Additional test data sets are needed to assess the best way to use the DP score for data obtained on perdeuterated proteins.
The RPF program can also analyze homodimeric proteins. This requires the user to first combine the two identical chains into a single chain with different residue index. A new chemical shift assignment table can be created based on the resonance assignments from the two identical chains, and match with the combined single chain residue index. We suspect that an accurate highly-degenerate homodimeric protein structure will require a higher DP score cutoff than a protein structure with less degenerate resonance frequencies.
2.2 Control Values
Edit/Review automated control values
Once you submit your input files, you will be prompted to review the default control values. For each peaklist, you should check that the columns are assigned correctly, and that the sweep width of that column is correct if there are folded peaks. You may set SW=1000 if there is no folded peak. You can also add a reference shift for any column and tweak the tolerances as appropriate.
A small preview of the peaklist will be visible to assist you.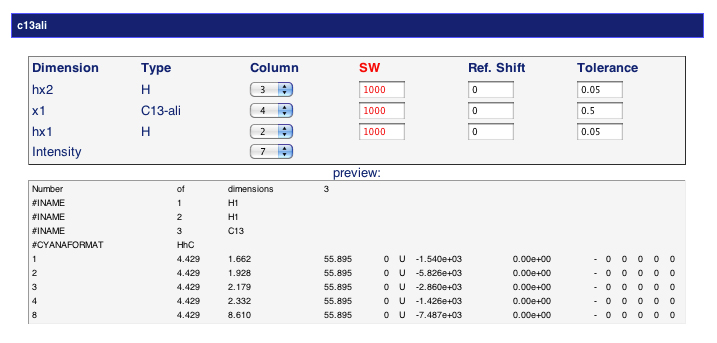
A small preview of the peaklist will be visible to assist you.
2.3 Calculation Results
View Calculation Results
The summary page reports the RPF and DP scores for multi-models. You can also view the RPF/DP scores for each model from the "Single-Models" page, and for each NOESY peak list (e.g. c13ali, n15noesy, c13aro pages). In this example, the DP score for the ensembles is 0.805.
The distribution of the false positive interactions (aka precision violations) is mapped on the query structure based on a heat index where red represents residue with strong precision violations, and blue represents residue with few or no precision violations. In this example, residues 29 and 32 are colored red, indicating some very short distances are observed between them based on the PDB structures, but the corresponding NOEs are not found in the data.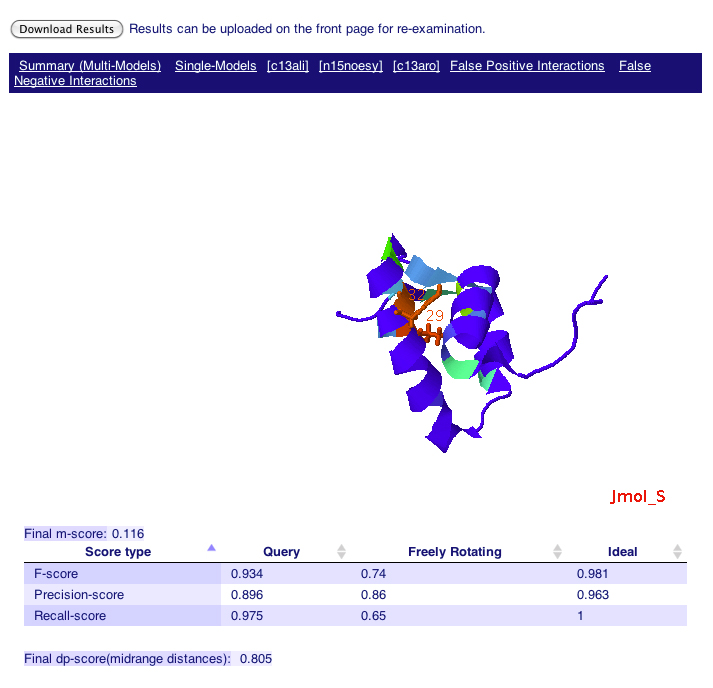
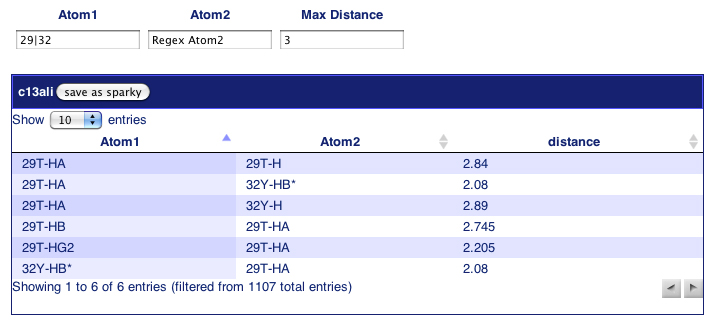
The "False Positive Interactions" page reports all distances (<= 5.0 ang) calculated from the query structures, but not supported by the NOE data. You may use regex to filter the list of those false positive interactions. In this example, there are only 6 violations from residue 29 or 32 with max distance of 3.0 ang.
The "False Negative Interactions" page reports observed peaks which are not supported by the query structures within the average distance of 5.0 ang.
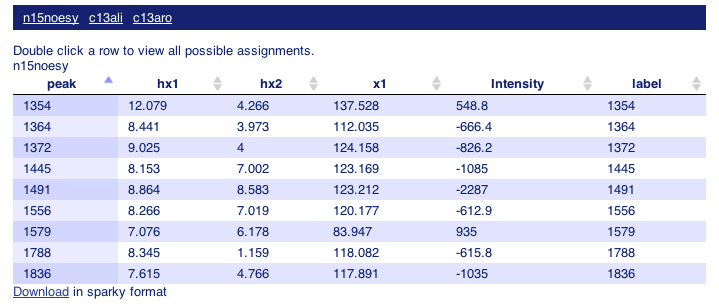
The "False Positive Interactions" and "False Negative Interactons" reports can be mapped to the spectrum for review and may provide guidance for further structure refinement.
The distribution of the false positive interactions (aka precision violations) is mapped on the query structure based on a heat index where red represents residue with strong precision violations, and blue represents residue with few or no precision violations. In this example, residues 29 and 32 are colored red, indicating some very short distances are observed between them based on the PDB structures, but the corresponding NOEs are not found in the data.
The "False Positive Interactions" page reports all distances (<= 5.0 ang) calculated from the query structures, but not supported by the NOE data. You may use regex to filter the list of those false positive interactions. In this example, there are only 6 violations from residue 29 or 32 with max distance of 3.0 ang.
The "False Negative Interactions" page reports observed peaks which are not supported by the query structures within the average distance of 5.0 ang.
The "False Positive Interactions" and "False Negative Interactons" reports can be mapped to the spectrum for review and may provide guidance for further structure refinement.
3. Web-Service (To be updated ...)
RPF Web-Service using SOAP with Attachments
The RPF Web-Service can be accessed by using the SOAP with Attachments protocol. Software Developers can directly consume the web-service using existing SOAP APIs.
Below are some examples of how to use the RPF web-service.
Below are some examples of how to use the RPF web-service.
3.1 Perl example
RPF Web-Service using SOAP::Lite
Perl developers can write SOAP web-service clients using the SOAP::Lite package, and can create SOAP attachments using the MIME::Entity package. Below is a sample script that demonstrates this.
code:
Download the example code
code:
#!/usr/bin/perl -w
#
#Sample script to demonstrate the use of the RPF Web-Service.
#It is important that the file attachments have the correct Content-Ids.
#
#accepted Ids are:
#Ids are bmrbfile, pdbfile, n15noesy, c13aro, c13ali, c13noesy, ccnoesy, and noesy2d
#
#required parameters are:
#email, protein_name, pdbfile, and bmrbfile
#
#optional parameters are:
# [experiment name]_water for the water suppression range
# [experiment name]_d20 if the experiment is in d20
#
use SOAP::Lite;
use MIME::Entity;
use SOAP::Lite::Packager;
sub build_entity {
my ($id, $filename, $filepath) = @_;
return build MIME::Entity
Type => "text/plain",
Path => $filepath,
Filename => $filename,
Disposition => "attachment",
Id => $id;
}
my $email = SOAP::Data->name(email => "gautams\@cabm.rutgers.edu")->type("string");
my $protein_name = SOAP::Data->name(protein_name => "str65")->type("string");
my $bmrb = &build_entity("bmrbfile", "str65.str", "/dataset/StR65/str65.str");
my $pdb = &build_entity("pdbfile", "StR65.pdb", "/dataset/StR65/StR65.pdb");
my $n15noesy = &build_entity("n15noesy", "n15noesy.list", "/dataset/StR65/n15noesy.list");
my $c13aro = &build_entity("c13aro", "c13aro.list", "/dataset/StR65/c13aro.list");
my $c13ali = &build_entity("c13ali", "c13ali.list", "/dataset/StR65/c13ali.list");
my $c13ali_water = SOAP::Data->name("c13ali_water" => "4.6-4.9")->type("string"); #water suppression range
my $c13ali_d20 = SOAP::Data->name("c13ali_d20"); #d20 solution
my $result = SOAP::Lite
-> service('http://nmr.cabm.rutgers.edu:80/rpf/RpfService.wsdl')
-> getRpfScoreWithOptions($email, $protein_name, $bmrb, $pdb, $c13aro, $c13ali, $n15noesy, $c13ali_water);
print $result . "\n";
Download the example code